

How To Establish Sample Sizes For Process Validation Using C=0 Sampling Plans
By Mark Durivage, Quality Systems Compliance LLC
The first article in this series, Risk-Based Approaches To Establishing Sample Sizes For Process Validation (June 2016), provided and established the relationship between risk and sample size. This article will demonstrate the use of C=0 sampling plans to establish sample sizes for process validation.
MIL-STD-105 – Sampling Procedures and Tables for Inspection by Attributes was officially cancelled in February 1995 and replaced by ANSI/ASQ Z1.4 Sampling Procedures and Tables for Inspection by Attributes. Nicholas Squeglia derived the Zero Acceptance Number Sampling Plans from the ANSI/ASQ Z1.4 standard, and his C=0 sampling plans greatly reduced the complexity associated with ANSI/ASQ Z1.4. For example, the use of normal, tightened, reduced inspection; switching rules; single sampling; and double sampling were not integrated in Squeglia’s procedure.
I would like to suggest that, whenever possible, variable sampling plans should be utilized — for two reasons. First, variables data will yield much more information than a comparable sample using attribute data. Secondly, with fewer samples, associated costs of inspection, measurement, and testing can be substantially reduced.
It should also be noted that a simple random sample is meant to be an unbiased representation of a group. A sampling error can occur with a simple random sample if the sample doesn’t end up accurately reflecting the population it is supposed to represent. In other words, each piece that is to be inspected, measured, or tested must have the same chance of being selected and being representative of the batch or lot.
Before we begin, we must establish our definitions of risk and their acceptable quality limit (AQL), the level of quality that will be accepted 95% of the time. These definitions can and should vary based upon the organizational needs. A good place to determine the risk level is from the failure mode and effects analysis (FMEA). FMEA (design, process, user) is a systematic group of activities designed to recognize, document, and evaluate the potential failure of a product or process and its effects. FMEA uses a risk priority number (RPN), which is comprised of frequency, detection, and severity. The higher the RPN, the higher the risk; however, a high severity in conjunction with low probability of occurrence and high probability of detection may still necessitate the appropriate controls for high risk. Table 1 depicts an example FMEA with the associated risk levels.
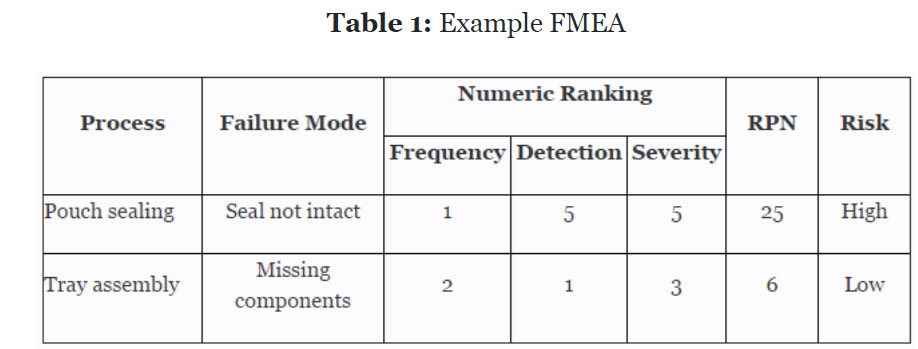
Once the risk level has been determined (low, medium, high), the appropriate AQL can be selected using Table 3. Figure 1 depicts the linkage from FMEA, risk, and AQL.

Table 2 shows an example of risk level definitions with accompanying defect classifications. These definitions can and will vary based upon the product(s) produced and their intended and unintended uses.
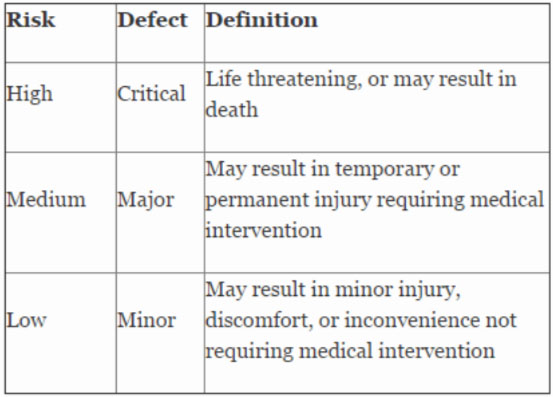
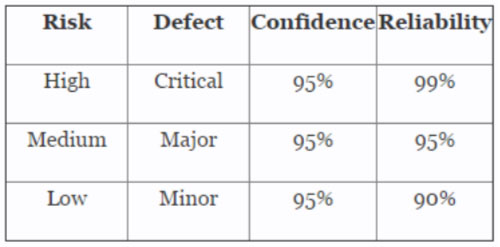
Table 3 depicts example AQLs based upon risk. Different AQLs can and should be utilized based upon the organizations risk acceptance determination threshold, industry practice, guidance documents, and regulatory requirements. A note of caution when using this method: Lot sizes used for validation activities should be consistent with the lot sizes anticipated for production.
Acceptance sampling is used to assess the quality based on sample size, acceptance number, and desired quality level. C=0 sampling plans are based on the premise of accepting the lot if zero defects are found during the inspection, and rejecting the lot if one or more defects are found during the inspection. Another way to define C=0 is the acceptance number (a) is zero (0); in other words, we accept the lot if zero (0) defects are detected. C=0 sampling plans provide more protection to the consumer, which is especially important when health and human welfare are involved. Using the C=0 sampling plan is the easiest method to determine a statistically valid sample size to support process validation activities, because all that is needed is the lot size (N) and a risk-based AQL.
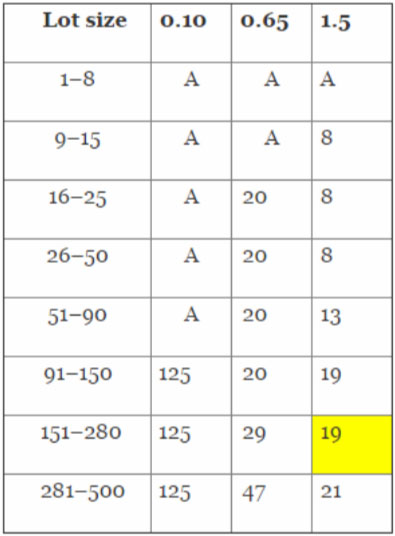
Note: ‘A’ means all pieces must be inspected.
A tray assembly process is determined to pose a low risk where a minor defect may result in minor injury, discomfort, or inconvenience not requiring medical intervention. According to Table 4, an AQL of 1.5 should be used. It is expected that a normal production run will consist of 250 pieces. With a lot size of 250 and an AQL of 1.50, 19 pieces must be randomly selected and inspected with zero (0) defects detected to meet the process validation acceptance criteria.
It should be noted that if the lot size were increased to 500 pieces, only an additional two (2) pieces would be required, a savings of 15 pieces were two (2) individual lots of 250 pieces manufactured. By strategically determining lot sizes, significant cost savings can be realized due to decreased inspection, measuring, and testing costs.
I want to reinforce that different AQLs and should be utilized based upon an organization’s risk acceptance determination threshold, industry practice, guidance documents, and regulatory requirements. Also realize that the smaller the AQL, the more inspection, measuring, and testing must be performed. Conversely, the larger the AQL, the less inspection, measuring, and testing is required.
Subsequent articles in this series will provide additional how-to examples on applying risk-based sample size techniques to process validations in your organization.
References:
- ANSI/ASQ Z1.4-2008: Sampling Procedures and Tables for Inspection by Attributes
- Durivage, M.A., 2014, Practical Engineering, Process, and Reliability Statistics, Milwaukee, ASQ Quality Press
- Durivage, M.A., and Mehta, B., 2016, Practical Process Validation, Milwaukee, ASQ Quality Press
- Durivage, M.A., 2016, Risk-Based Approaches To Establishing Sample Sizes For Process Validation, Life Science Connect
- Squeglia, Nicholas K. 2008, Zero Acceptance Number Sampling Plans. 5th ed. Milwaukee: ASQ Quality Press.
About The Author:
 Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC and is an ASQ Fellow and SRE Fellow. He earned a B.A.S. in computer-aided machining from Siena Heights University and an M.S. in quality management from Eastern Michigan University. He holds several certifications including CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments, and connect with him on LinkedIn.
Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC and is an ASQ Fellow and SRE Fellow. He earned a B.A.S. in computer-aided machining from Siena Heights University and an M.S. in quality management from Eastern Michigan University. He holds several certifications including CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments, and connect with him on LinkedIn.
Article originally published on September 7, 2016, in Pharmaceutical Online